All News
UMIACS Expands Data Center to Support Research Growth

Jisha Jesudass (right in photo) is responsible for designing and maintaining a sophisticated network system used by UMIACS faculty to seamlessly move data among various stakeholders both on and off campus.
For almost four decades, a powerful data center in the A.V. Williams Building has been an essential part of the University of Maryland Institute for Advanced Computer Studies (UMIACS), supporting the institute’s mission of incentivizing cutting-edge research and staying at the forefront of technological innovation.
As the institute’s research portfolio continues to expand, the infrastructure needed to support it—particularly in compute-heavy areas like artificial intelligence (AI) and machine learning—also needs to grow.
In April, UMIACS data center staff began coordinating a three-month renovation to expand the center’s physical footprint by 50 percent, growing from 2,300 square feet to 3,450 square feet.
The expansion project will provide additional space for computing hardware and will offer better options to ensure that the data center’s power grid and cooling systems can handle any increased workload, says Derek Yarnell, the institute’s director of computing facilities.
Much of the current workload is handled by multiple racks of graphical processing units (GPUs), powerful parallel computing hardware used for gaming, content creation—and in the case of UMIACS—machine learning and neural networks for high performance computing in areas like AI and computer vision.
In the past five years, UMIACS has increased the number of GPUs in its data center by almost tenfold, Yarnell says. This has allowed researchers to perform certain tasks at almost one petaFLOP of speed—equivalent to more than one quadrillion floating-point operations per second.
“If you measure the amount of computational infrastructure in place that is dedicated specifically toward AI and machine learning research, we are the largest on campus by a wide margin,” Yarnell says. “And we believe this is only the beginning.”
UMIACS faculty are constantly seeking new mechanisms to upgrade their computing resources, says Tom Goldstein, a professor of computer science and director of the University of Maryland Center for Machine Learning (CML).
Goldstein advocates for making economy of scale purchases, wherein CML faculty collectively pool portions of their individual research grants to purchase racks, routers, wiring and accessories needed to support multiple GPU clusters.
The data center expansion matches well with that concept.
“We want to be forward-looking in regard to increasing our computing capabilities, particularly those of us requiring high-memory nodes for projects that focus on large language models and other emerging AI research,” Goldstein says.
Other UMIACS faculty also want to improve both the availability and level of computing power available through the institute’s data center.
Furong Huang, an assistant professor of computer science with an appointment in UMIACS, is working with her graduate students to build a foundation model for sequential decision making, a form of generalist AI that holds the capacity to learn and master diverse sets of tasks downstream.
“Imagine an AI that not only learns quickly, but adapts to your daily tasks autonomously,” says Huang, who is a member of CML.
This work requires analyzing massive amounts of data used in the AI system, a task best handled by a large network of multiple GPUs, Huang says.
Supporting these myriad research efforts falls on Yarnell’s team, a dedicated staff of network engineers and high-performance computing experts assisted by more than 20 undergraduates working at the UMIACS Help Desk.
The institute’s technical staff are involved in the computing process from start to finish, Yarnell says. This includes helping faculty spec out their equipment needs to submit in their research proposals; working with UMD procurement to purchase the equipment; installing all the hardware and software when it arrives on campus; and maintaining the equipment over its technical lifespan by providing security patches and updates.
The tech staff is also responsible for managing and securely storing all the data used in UMIACS research. Current storage capabilities top out at around 3.65 petabytes, with that number constantly increasing, says UMIACS network engineer Jisha Jesudass.
There has also been steady growth in the network capabilities for the institute. When UMIACS moved from the A.V. Williams Building to the Brendan Iribe Center for Computer Science and Engineering in 2019, Jesudass’ job responsibilities included designing and maintaining a high-capacity network—miles of wiring and dozens of sophisticated routers—used to transmit large volumes of data both on and off campus.
She emphasized that the upgrades currently underway in the data center include further improving connectivity capabilities to make the workflow of UMIACS projects more efficient.
“Every job or every researcher right now, they want to minimize latency and delays. This new infrastructure will help a lot with that,” Jesudass says.
 For Yarnell (pictured left), the renovation and expansion of the data center remains a work in progress and a labor of love. His involvement with the data center started in 1998 as an undergraduate working at the UMIACS Help Desk. Since that time, he’s seen—and helped lead—much of the data center’s growth.
For Yarnell (pictured left), the renovation and expansion of the data center remains a work in progress and a labor of love. His involvement with the data center started in 1998 as an undergraduate working at the UMIACS Help Desk. Since that time, he’s seen—and helped lead—much of the data center’s growth.
“Our data center has obviously stood the test of time up to this point—it’s like a Ferrari, a fine-tuned machine that has aged very well,” he says. “But we still think we can continue to innovate it, and continue to build it, in a way that will allow the center to support any new opportunities that are yet to come.”
—Reporting on this story by Shaun Chornobroff, UMIACS communications group
University of Maryland Hosts Microbiome Research Symposium

More than 80 people braved stark wintry conditions on January 16 to attend a research symposium at the University of Maryland that explored the world of complex microbial communities.
The Symposium on Microbiome Research at the Interface of Environment, Health and Agriculture joined researchers from the federal government, academia and private industry who are focused on the connectivity between microbes interacting with each other, the environment, agricultural systems, and human and animal health.
Hosted by the University of Maryland Center of Excellence in Microbiome Sciences, the event featured multiple talks, breakout sessions, an engaging poster session, and a networking reception. All the events went off without a hitch, despite 4 inches of snow that closed the university for the day and made travel difficult.
“We were fortunate that more than two-thirds of the people who registered were able to show up and participate,” says Mihai Pop, a UMD professor of computer science who is the director of the microbiome center. “We were particularly pleased by the strong turnout from federal scientists in the region, as well as colleagues from the medical and dentistry schools in Baltimore.”
A morning keynote talk by Susannah Tringe, division director of the DOE Joint Genome Institute at the Lawrence Berkeley National Laboratory, looked at the sequence-based interrogation of soil microbiomes, and how those microbes can benefit various ecosystems.
The afternoon keynote by Joff Silberg, a professor of biosciences at Rice University, was presented virtually as Silberg was unable to fly out of Houston due to poor weather. His talk explored the use of engineered living microbes, and how they might be used to monitor various soil pollutants in real time.
Other talks included how microbial communities can impact coffee growers, the effect of cow manure microbes on farm soil, microbial activity related to women’s gynecologic health, and other topics focused on human gut bacteria and inflammatory bowel disease.
“There’s such a rich diversity of perspectives and ongoing work at the University of Maryland involving microbiome sciences,” says Hannah Zierden, an assistant professor of chemical and biomolecular engineering at UMD and core member of the microbiome center. “I’m excited at the opportunities we have and look forward to continued collaborations—as well as new ones—as we expand our outreach and impact.”
Zierden presented some recent research from her own UMD lab at the conference, which aims to better understand the function of bacterial extracellular vesicles produced by vaginal microbes, and how they might be used to engineer biocompatible therapies for healthy pregnancies.
A large contingent of researchers from the University of Maryland School of Dentistry were onsite for the symposium, including Areej Alfaifi, a doctoral student in the dental biomedical sciences program.
“This event broadened my perspective by introducing me to entirely different aspects of microbiome studies,” says Alfaifi, whose dissertation explores the use of genomic sequencing tools to gain a deeper understanding of the oral microbiome in COVID-19 patients. “Connecting with students and faculty from different schools was an amazing experience that reshaped my thoughts on the field. This meeting was truly unforgettable!”
Additional attendees included faculty, postdocs and graduate students from the University of Delaware, Towson University, University of Maryland School of Medicine, and the University of Maryland, College Park.
Federal scientists in attendance hailed from the USDA, FDA, Department of Energy, and the Smithsonian National Zoo, with representatives from QIAGEN, CosmosID—both major sponsors of the symposium—also present.
The symposium also received support from the University of Maryland Institute for Advanced Computer Studies, Mid-Atlantic Microbiome Meet-up, and the UMD’s Grand Challenges Grants program.
Pop said the UMD microbiome center will help coordinate another symposium in 2025 in Baltimore, working closely with the Institute for Genome Sciences at the University of Maryland, Baltimore to investigate new topics related to microbiome sciences.
“We expect to continue our momentum in this area, which reaches across multiple scientific, medical and policy-related disciplines,” Pop says. “Our belief is that the basic unresolved questions involving microbial communities are interrelated—and so are the solutions we’re working on.”
—Story by Maria Herd, UMIACS communications group
What Makes Urine Yellow? UMD Scientists Discover the Enzyme Responsible
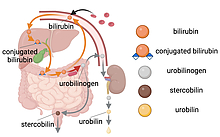
Researchers at the University of Maryland and National Institutes of Health have identified the microbial enzyme responsible for giving urine its yellow hue, according to a new study published in the journal Nature Microbiology on January 3, 2024.
The discovery of this enzyme, called bilirubin reductase, paves the way for further research into the gut microbiome’s role in ailments like jaundice and inflammatory bowel disease.
“This enzyme discovery finally unravels the mystery behind urine’s yellow color,” said the study’s lead author Brantley Hall, an assistant professor in the University of Maryland’s Department of Cell Biology and Molecular Genetics. “It’s remarkable that an everyday biological phenomenon went unexplained for so long, and our team is excited to be able to explain it.”
When red blood cells degrade after their six-month lifespan, a bright orange pigment called bilirubin is produced as a byproduct. Bilirubin is typically secreted into the gut, where it is destined for excretion but can also be partially reabsorbed. Excess reabsorption can lead to a buildup of bilirubin in the blood and can cause jaundice—a condition that leads to the yellowing of the skin and eyes. Once in the gut, the resident flora can convert bilirubin into other molecules.
“Gut microbes encode the enzyme bilirubin reductase that converts bilirubin into a colorless byproduct called urobilinogen,” explained Hall, who has a joint appointment in the University of Maryland Institute for Advanced Computer Studies and is a core faculty member in the Center for Bioinformatics and Computational Biology. “Urobilinogen then spontaneously degrades into a molecule called urobilin, which is responsible for the yellow color we are all familiar with.”
Urobilin has long been linked to urine’s yellow hue, but the research team’s discovery of the enzyme responsible answers a question that has eluded scientists for over a century.
Aside from solving a scientific mystery, these findings could have important health implications. The research team found that bilirubin reductase is present in almost all healthy adults but is often missing from newborns and individuals with inflammatory bowel disease. They hypothesize that the absence of bilirubin reductase may contribute to infant jaundice and the formation of pigmented gallstones.
“Now that we’ve identified this enzyme, we can start investigating how the bacteria in our gut impact circulating bilirubin levels and related health conditions like jaundice,” said study co-author and NIH Investigator Xiaofang Jiang. “This discovery lays the foundation for understanding the gut-liver axis.”
In addition to jaundice and inflammatory bowel disease, the gut microbiome has been linked to various diseases and conditions, from allergies to arthritis to psoriasis. This latest discovery brings researchers closer to achieving a holistic understanding of the gut microbiome’s role in human health.
“The multidisciplinary approach we were able to implement—thanks to the collaboration between our labs—was key to solving the physiological puzzle of why our urine appears yellow,” Hall said. “It’s the culmination of many years of work by our team and highlights yet another reason why our gut microbiome is so vital to human health.”
###
This article was adapted from text provided by Brantley Hall and Sophia Levy.
In addition to Hall, UMD-affiliated co-authors included Stephenie Abeysinghe (B.S. ’23, public health science); Domenick Braccia (Ph.D. ’22, biological sciences); biological sciences major Maggie Grant; biochemistry Ph.D. student Conor Jenkins; biological sciences Ph.D. students Gabriela Arp (B.S. ’19, public health science; B.A. ’19, Spanish language), Madison Jermain, Sophia Levy (B.S. ’19, chemical engineering; B.S. ’19, biological sciences) and Chih Hao Wu (B.S. ’21, biological sciences); Glory Minabou Ndjite (B.S. ’22, public health science); and Ashley Weiss (B.S. ’22, biological sciences).
Their paper, “Discovery of a gut microbial enzyme that reduces bilirubin to urobilinogen,” was published in the journal Nature Microbiology on January 3, 2024.
This research was supported by the NIH’s Intramural Research Program, the National Library of Medicine and startup funding from UMD. This article does not necessarily reflect the views of these organizations.
Colwell Honored with Builders of Science Award from Research!America

A noted University of Maryland researcher is being honored for their pioneering insights into microbial water quality surveillance and longstanding efforts in fighting waterborne diseases on a global scale.
Distinguished University Professor Rita Colwell was recently named a recipient of the 2024 Builders of Science Award, one of the top Advocacy Awards given each year by Research!America, a nonprofit alliance that supports increased funding and better policies related to medical and health research.
The Builders of Science Award is specific to those who have provided leadership and determination in building an outstanding scientific research organization, as well as those who have been at the forefront of scientific research.
Still active in research through her appointment in the University of Maryland Institute for Advanced Computer Studies (UMIACS), much of Colwell’s scientific career has been focused on tracking and predicting outbreaks of dangerous waterborne pathogens by combining bioinformatics with satellite imaging. She recently published a report that identified several dangerous bacteria in Florida’s coastal waters following Hurricane Ian in 2022.
The impact of Colwell's research and scholarship has been especially felt in cholera-endemic countries worldwide—the result of a key discovery she made in the 1970s involving cholera-causing bacteria, known as Vibrio cholera. Previously thought to be incapable of surviving more than a few hours outside a human host, she discovered that the bacteria occur naturally in the aquatic environment associated with plankton.
This highlighted the critical link between the environment and cholera and led to Colwell's subsequent application of satellite imagery and modelling to predict cholera outbreaks, as well as her innovative use of sari cloths as filters to greatly reduce contamination in drinking water.
Cowell was the 11th director and first woman to lead the National Science Foundation, and during her tenure (1998–2004), she oversaw its most significant period of growth. She also championed and secured NSF funding for innovative science and engineering education programs and initiatives to advance women in academic engineering and science careers.
Other notable awards and recognition that Colwell has received include the National Medal of Science; the Stockholm Water Prize; membership into the National Academy of Sciences, Royal Society of Canada, Swedish Royal Academy of Science, Irish Royal Academy of Science, and the Bangladesh and Indian academies of science; and the “The Order of the Rising Sun, Gold and Silver Star” from the emperor of Japan.
Colwell and the other Advocacy Award recipients will be officially recognized on March 13, 2024 at an event at the National Academy of Sciences in Washington, D.C.
New Study Confirms Presence of Dangerous Bacteria in Florida’s Coastal Waters Following Hurricane Ian

Caption: Hurricane Ian is pictured from the International Space Station as it orbited 258 miles above the Caribbean Sea on September 26, 2022. At the time of this photograph, Ian was just south of Cuba gaining strength and heading toward Florida.
When Hurricane Ian struck southwest Florida in September 2022, it unleashed a variety of Vibrio bacteria that can cause illness and death in humans, according to a new study published in the journal mBio.
Using a combination of genome sequencing and satellite and environmental data, a team of researchers from the University of Maryland, the University of Florida and microbiome company EzBiome detected several pathogenic Vibrio species in water and oyster samples from Florida’s Lee County, a coastal region that was devastated by Hurricane Ian. The samples, which were collected in October 2022, revealed the presence of two particularly concerning species: Vibrio parahaemolyticus and Vibrio vulnificus.
“We were very surprised to be able to detect—without any difficulty—the presence of these pathogens,” said the study’s senior author Rita Colwell, a Distinguished University Professor in the University of Maryland Institute for Advanced Computer Studies (UMIACS) who has studied Vibrio for the last 50 years.
The study’s findings correspond with a reported increase in V. vulnificus cases in the state of Florida in October 2022. According to the Florida Department of Health, Lee County—which had the highest caseload in the state—reported 38 infections and 11 deaths linked to vibriosis.
Vibrio bacteria naturally occur in the ocean, where they live symbiotically with crustaceans, zooplankton and bivalves. When the bacteria encounter humans, some species can cause an infection known as vibriosis, but the side effects depend on the type of Vibrio and severity of the infection. V. parahaemolyticus can cause gastroenteritis and wound infections, while the V. vulnificus species can cause necrotizing fasciitis—a flesh-eating infection—and kills 1 in 5 infected people.
People can contract vibriosis by eating raw or undercooked seafood or by getting seawater in an open wound. Because Vibrio thrive in warm saltwater, hurricanes and floods can increase the chances of a person becoming exposed.
Several conditions during and after Hurricane Ian favored the growth of Vibrio bacteria, including the amount of rainfall, changes in sea surface temperature and concentrations of chlorophyll in the ocean, which can indicate densities of phytoplankton—and subsequently zooplankton—in an area. In places with plankton blooms, the researchers found an abundance of Vibrio bacteria.
With warming oceans expected to fuel wetter and more powerful storms like Ian, coastal communities could see more Vibrio infections in the future.
“These Vibrios generally grow well between 15 and 40 degrees Celsius [59–104 degrees Fahrenheit], so as the temperature warms, their generation time shortens and they divide faster and faster,” Colwell said. “The warming of seawater—which mixes with freshwater, creating optimal salinities—really enhances the growth of Vibrios, so it’s a very serious concern.”
While the environmental conditions in Florida following Hurricane Ian were ripe for vibriosis, these cases are not limited to southern climes. In August 2023, three people in New York and Connecticut died from V. vulnificus infections.
Colwell and her co-authors—which included Kyle Brumfield (Ph.D. ’23, marine estuarine environmental sciences) and UMD Cell Biology and Molecular Genetics Research Professor Anwar Huq—predicted this recent spike in vibriosis cases based on trending environmental conditions in the Northeast United States. As ocean temperatures continue to rise, Colwell said the rapidly warming Chesapeake Bay could also be affected.
“The waters are much warmer in Florida right now than they are in the Chesapeake Bay, but on a lot of the East Coast, the waters are warming,” Colwell said. “This is a threatening indication that we may be seeing more Vibrio vulnificus infections.
Colwell and her co-authors noted that while they analyzed only a limited number of samples, their findings illustrate the potential of genetic analysis, environmental data and remote sensing to improve public health by proactively detecting and characterizing Vibrio pathogens.
They also called for further investigation to quantify the prevalence of Vibrio bacteria in different locations, seasons and environmental conditions. Colwell said this research is not only vital to public health but also an important step in understanding our changing climate.
“On the positive side, knowing that these infections are associated with the increased variability of a changing climate, perhaps now is the time to develop mechanisms to understand and mitigate it,” Colwell said. “Climate change and flooding are clearly linked to infectious disease, and we need to take it seriously.”
###
The paper “Genomic Diversity of Vibrio spp. and Metagenomic Analysis of Pathogens in Florida Gulf Coastal Waters Following Hurricane Ian” was published in mBio on October 16, 2023.
—Story by Emily Nunez, College of Computer, Mathematical, and Natural Sciences communications team
CBCB Researchers Present Four Papers at Prestigious Algorithms in Bioinformatics Workshop

Erin Molloy (left in photo), an assistant professor of computer science, and doctoral student Tobias Rubel (right), are part of a team of University of Maryland researchers presenting their work at WABI 2023.
Researchers from the Center for Bioinformatics and Computational Biology (CBCB) are presenting four papers this week at the Workshop on Algorithms in Bioinformatics (WABI 2023). The annual conference—held this year from September 4–6 in Houston, Texas—brings together experts from around the world who are focused on discrete algorithms and machine-learning methods that address important problems in molecular biology.
The UMD researchers are discussing work that includes a new algorithm for the parallel construction of suffix arrays—one of the key tools for processing vast amounts of genomic data—and other research that explores novel computational methods to study the human genome and the evolution of tumors.
“WABI is a very important conference in our field, and has a rich history of impactful papers,” says Michael Cummings, a professor of biology with an appointment in the University of Maryland Institute for Advanced Computer Studies (UMIACS) who is the director of CBCB. “The four papers in this conference are evidence of the quality of research from our faculty and students.”
One of the papers, “Fast, Parallel, and Cache-friendly Suffix Array Construction,” offers a new approach for building suffix arrays that coalesce efficiently with modern CPUs.
The suffix array is a classic data structure used to “index” sequences. That is, given some very long sequence, like a genome, the suffix array allows scientists to search for a particular query fast. This data structure can tell them if the query occurs and how many times, as well as the precise location in the underlying sequence.
Due to its importance, algorithms for building the suffix array have been studied for decades, says Jamshed Khan, a fifth-year computer science doctoral student who is lead author on the paper.
In addition to answering questions about how much computation is necessary to construct the suffix array, the problem has also been examined from the perspective of parallel algorithms. Specifically, if researchers have access to processors with many cores, how can they make use of this capability of parallel computation to speed up the suffix array construction?
Khan says the algorithm developed by the CBCB team is conceptually simpler than several existing parallel algorithms. “We demonstrate that when parallel algorithms start outperforming the fastest serial algorithms, ours is the fastest, and that it scales better as more parallel cores are made available,” he says.
Joining Khan in this work are co-authors Rob Patro, an associate professor of computer science, Laxman Dhulipala and Erin Molloy, both assistant professors of computer science, and Tobias Rubel, a third-year doctoral student in computer science.
Patro, Dhulipala and Molloy all have joint appointments in UMIACS.
Another paper presented at the workshop, “Fulgor: A Fast and Compact k-mer Index for Large-Scale Matching and Color Queries,” defines and develops a new generation of efficient and modular indexes for k-mer queries—short sequences of DNA and RNA with a fixed length.
“Specifically, our k-mer color query tells us which reference sequences contain a queried k-mer, but not where said k-mer occurs,” says lead author Jason Fan, who just defended his Ph.D. dissertation in computer science.
This would be like rapidly searching for a particular word in a set of books and articles and knowing it’s there but not exactly where, Fan explains.
However, this approximation is sufficient for useful computational analyses—such as taxonomic classification—and, more importantly, enables the analysis and indexing of massive collections of reference genome sequences.
“We demonstrate in our experiments that it is at least two times smaller than the prior state-of-the-art methods and at least twice as fast,” Fan says. “Excitingly, we have already begun to improve and develop new and more efficient algorithms that better exploit the structure of indexed data.”
In addition to Fan, co-authors of the paper are Rob Patro, Jamshed Khan, Noor Pratap Singh, a fifth-year doctoral student in computer science, and Giulio Ermanno Pibiri, an assistant professor of computer science at Ca’Foscari University of Venice.
Molloy and her students are presenting two additional papers at WABI 2023. The first, “Quartets Enable Statistically Consistent Estimation of Cell Lineage Trees Under an Unbiased Error and Missingness Model,” expands upon what researchers know about models of cancer evolution, proposing a new methodology for reconstructing the evolutionary history of a tumor.
Tumor phylogenies, as they are called, reveal how cells in a tumor evolved from a common ancestor cell through cell division. Molloy and lead author Yunheng Han, a fifth-year doctoral student in computer science, say that accurate reconstruction is critical for studying cancer progression and response to treatment, and ultimately the development of more effective therapies. Their new method is a promising path forward given its strong statistical properties.
The second paper, “Leveraging Constraints Plus Dynamic Programming for the Large Dollo Parsimony Problem,” presents a new algorithmic approach to the Dollo parsimony problem, which has the goal of finding the simplest evolutionary tree (phylogeny) that explains the data, under the assumption that mutations are rare and thus are unlikely to occur multiple times across the tree.
In contrast, the loss of the mutation is more likely and as such could reasonably occur multiple times. These assumptions hold true for tumor evolution and some data types for species evolution. The researchers implemented and evaluated their algorithm in the context of the latter—using both simulated and real data sets—finding that it reliably improves the performance of prior methods.
Future work will evaluate the utility of the method in the context of cancer.
Junyan Dai, a senior double majoring in mathematics and computer science, was the lead author of the paper, teaming up with Rubel, Han and Molloy.
—Story by Melissa Brachfeld, UMIACS communications group
REU Interns Learn to Navigate Complexities of Health Research

The students were part of the highly competitive Bioinformatics Research In Data science for Genomics (BRIDGE), a program that’s supported by the National Science Foundation’s Research Experiences for Undergraduates (REU).
Run by faculty and graduate students in the Center for Bioinformatics and Computational Biology (CBCB), the 10-week program is an opportunity for students to tackle their research questions in the field for the first time, says Michael Cummings, a professor of biology and director of the center.
“It may result in a research publication, and allows them to work in a setting that is just focused on research,” he adds, noting that this year’s cohort of REU students had a 5% acceptance rate.
Sarah Blankespoor, a rising senior at the California Polytechnic State University majoring in public health, is working on genome polishing with neural networks while being advised by Mihai Pop, a professor of computer science and director of the University of Maryland Institute for Advanced Computer Studies (UMIACS).
Blankespoor says the goal of this research is to improve the accuracy of pathogen and mutation identification. She and Pop hope this technology will be used to diagnose pathogens and identify different mutations that occur in the gut microbiome.
Maddie Bonanno, a rising senior at Tulane University, is majoring in computer science and cell and molecular biology with a minor in mathematics. This summer, Bonanno was advised by Erin Molloy, an assistant professor of computer science.
Their research focused on attempting to find the best software to sort the evolutionary history of organism groups. Such analyses are called phylogenetics and can be applied to the DNA or amino-acid sequence of genes.
In addition to their research projects, Cummings says the students learned how to deal with general issues, such as time management, communication, problem specification and facing setbacks. He notes that these lessons expose students to what higher level research looks like.
Tobias Rubel, a third-year computer science Ph.D. student, served as a mentor to this year’s interns. They say the program allows students to expand their research abilities and see if they want to pursue a research career.
For some students, their summer projects will continue on. Both Bonanno and Blankespoor, along with their respective advisers, say they will keep investigating their respective topics because the program helped them develop a deep passion for research.
—Story by Ethan Cannistra, UMIACS communications group
USM Dedicates Rita Rossi Colwell Center in Baltimore

(From left) University of Maryland President Darryll J. Pines, Distinguished University Professor Rita R. Colwell, and Mihai Pop, director of the University of Maryland Institute for Advanced Computer Studies, where Colwell is active as a computational biologist.
A Baltimore building housing the University System of Maryland (USM) headquarters and an institute focused on marine and environmental technology now bears the name of a University of Maryland pioneering microbiologist.
The Rita Rossi Colwell Center honors the distinguished university professor renowned for her work combatting waterborne diseases and her advocacy for increasing the number of women in science and technology.
“It is truly an honor to be able to come to work in the Rita Rossi Colwell Center,” USM Chancellor Jay A. Perman, M.D., said at a naming celebration on June 22. “We had a unique opportunity to name a building after one of our own—a pioneering woman scientist and scholar who’s dedicated her life and career to improving human and environmental health, which we all know are inextricably linked.”
University of Maryland President Darryll J. Pines told an audience of more than 200 academic and regional leaders that as a young scientist he received his National Science Foundation (NSF) CAREER Award when Colwell was its director. Other speakers at the ceremony included University of Maryland, Baltimore County President Valerie Sheares Ashby and current NSF Director Sethuraman Panchanathan.
In December, the USM Board of Regents voted to rename the facility for Colwell, who was instrumental in founding the Columbus Center—as the building was originally known—with the goal of expanding environmental science education and research in Baltimore.
Colwell joined UMD in 1972 as a tenured professor and later assumed—sometimes concurrently—academic administrative appointments at UMD and within the university system, including at the University of Maryland Sea Grant College, Center for Environmental and Estuarine Studies, UMD’s Division of Academic Affairs, and the Center of Marine Biotechnology and the Maryland Biotechnology Institute.
Cowell was the 11th director and first woman to lead the NSF, and during her tenure (1998–2004), she oversaw its most significant period of growth. She also championed and secured NSF funding for innovative science and engineering education programs and initiatives to advance women in academic engineering and science careers.
Still active in research through her appointment in the University of Maryland Institute for Advanced Computer Studies, Colwell tracks and predicts outbreaks of cholera by combining bioinformatics with satellite imaging.
Notable awards and recognition that Colwell has received include the National Medal of Science; the Stockholm Water Prize; membership into the National Academy of Sciences, Royal Society of Canada, Swedish Royal Academy of Science, Irish Royal Academy of Science, and the Bangladesh and Indian academies of science; and the “The Order of the Rising Sun, Gold and Silver Star” from the emperor of Japan.
Cummings Awarded NIH Grant to Inform Malaria Vaccine Design
Despite decades of eradication efforts, malaria remains a global cause of disease and death, with the greatest burden largely affecting young children in Sub-Saharan Africa.
In 2021, an estimated 247 million people worldwide were infected with malaria resulting in 619,000 deaths, according to the World Health Organization (WHO).
That same year, the WHO approved the first malaria vaccine which has shown to significantly reduce deaths among young children. However, compared to other adolescent vaccinations, it has modest efficacy, preventing only about 30 percent of severe malaria cases.
Supported by a $483,000 grant from the National Institutes of Health (NIH), Michael Cummings, a professor of biology with an appointment in the University of Maryland Institute for Advanced Computer Studies, is working to understand the immune response to malaria to help scientists develop more effective vaccines.
Along with Cummings, who brings extensive biological data science expertise, the research team is comprised of Assistant Professor Andrea Berry, a pediatric infectious disease physician and malaria expert at the University of Maryland School of Medicine, and Assistant Professor John Altin, an immunology and genomics expert at the Translational Genomics Research Institute.
Their research investigates immune responses to malaria infection in infants and children in the Sub-Saharan countries Mali and Malawi, which are geographically distant sites with different malaria transmission patterns.
Cummings, who is also the director of the Center for Bioinformatics and Computational Biology, says the bulk of the data will be collected using a new customizable lab platform called PepSeq. The platform, which Altin helped create, starts with designing a “library” of peptides of interest—short strings of amino acids that are the building blocks of proteins. Each peptide is then linked to a unique DNA tag, which allows scientists to pinpoint which peptides are being targeted by which antibodies (or other proteins) in a sample.
The team will use a combination of statistical and machine learning approaches to analyze hundreds of thousands of peptides, with the goal of identifying antigens that will help inform vaccine development.
—Story by Melissa Brachfeld, UMIACS communications group
UMD Researchers Partner with Wilmer Eye Institute to Enhance Sustained Delivery of Ocular Drugs
Computational biologists at the University of Maryland are partnering with ophthalmologists from the renowned Wilmer Eye Institute at John Hopkins University to improve therapeutic options for people suffering from chronic ocular diseases.
Michael Cummings (left in photo) a professor of biology with an appointment in the University of Maryland Institute for Advanced Computer Studies, and Renee Ti Chou (right), a fourth-year computational biology doctoral student, are using machine learning algorithms and other methods to enhance how certain drugs are administered to patients suffering from serious eye ailments.
Their research is covered in-depth in a paper published on May 2, 2023, in the prestigious journal Nature Communications.
According to the Centers for Disease Control and Prevention, more than 4.2 million Americans aged 40 years and older are either legally blind or have low vision, primarily due to age-related eye diseases such as macular degeneration, cataracts, diabetic retinopathy, and glaucoma.
Surgery is an option for some, but for many others, treatment involves strictly adhering to eyedrop dosing regimens or frequent intraocular injections. This has proven to be problematic, with studies showing that only 64% of patients being treated for glaucoma adhere to the three-times daily eyedrop dosing schedule over a four-week period.
Implantable drug delivery systems remain a promising option for the sustained, controlled delivery of therapeutics in the eye, but they come with their own set of risks, including adverse reactions to the cornea and other side effects.
The UMD researchers, working with ophthalmologists, bioengineers, and pharmacologists at The John Hopkins University School of Medicine—home to the Wilmer Eye Institute—have an alternative solution, one that involves a process known as peptide engineering.
Chou, who is the co-lead author of the paper, says the key to their work is to improve the drug binding properties in ocular melanin, which is found in pigmented eye tissues and protects the eye from harmful ultraviolent light. Ocular melanin can act as a sustained-release depot in the eye for drug therapeutics, Chou adds, allowing the drugs to remain active for longer periods, which translates to less eyedrops or intraocular injections.
But not all drugs bind well to melanin, so the researchers are hoping to use peptides, which are short sequences of amino acids, as an “adaptor” to impart the melanin binding property to non-melanin binding drugs.
“In our study, we linked the candidate peptide to an ocular drug through conjugation and demonstrated that it can increase the drug retention time from eight days to up to 18 days, using an intraocular pressure reducing drug called brimonidine as an example,” Chou says.
Cummings, who is director of the Center for Bioinformatics and Computational Biology, says they used machine learning-based analytical methodology to develop the peptide adaptor, which is able to engineer new types of peptides that have a high cell-penetrating ability coupled with low cytotoxicity properties.
“We believe this method will have a distinct advantage over current implantable devices, as it does not require surgery or device removal, and is expected to have fewer side effects, both of which can improve a patient’s quality of life,” says Chou.
—Story by Melissa Brachfeld, UMIACS communications group